
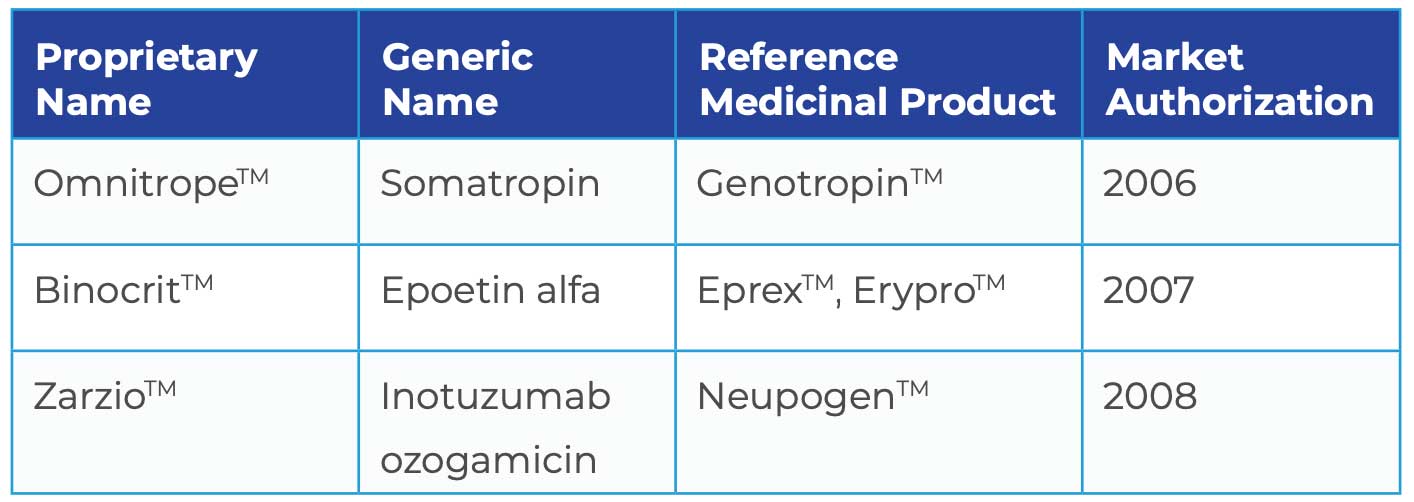
Novartis’ Sandoz unit pioneered the global biosimilar market with the development of a follow-on version of GenotropinTM which is currently marketed in the EU, US, Australia and Japan. By now, Sandoz has marketed three biosimilars in Europe indicating that the EU has sprinted ahead of the US in its approval of biosimilars:
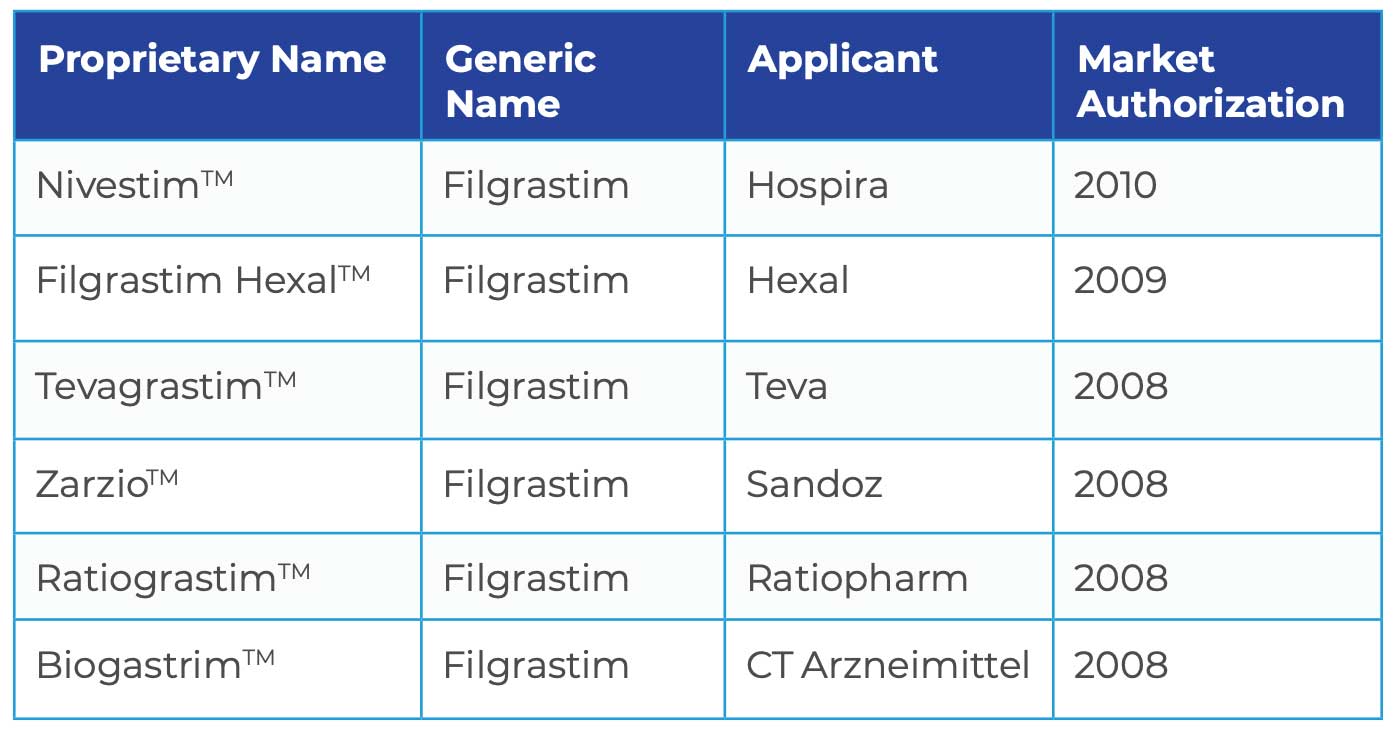
To appreciate the dynamics of the biosimilar market in Europe, there are currently six approved biosimilars to Amgen’s Neupogen™.
Without an abbreviated pathway for the approval of ‘generic’ versions of biologics in the US, the first generation of existing versions of similar biologics such as erythropoietin (ProcritTM and EpogenTM), botulinum toxin (BotoxTM, DysportTM, and MyoblocTM) and filgrastim (NeutrovalTM) were approved under a full Biosimilar License Agreement (BLA) each of which is not deemed interchangeable with its reference medicinal product. With the passage of the Biologics Price Competition and Innovation Act (‘the Biosimilar Act’) in 2009, an abbreviated pathway has also been created in the US for what is intended to be a streamlined development and FDA approval pathway for competing versions of already-marketed biologic drug products. Approval as a ‘biosimilar’ under the Biosimilar Act should save sponsors time and money in the development and approval process and must provide overall cost savings to patients and the health care system.
Although the new Biosimilar Act has been subject to criticism for its complexity with many important details left undefined or open-ended, it seems to have spurred many pharmaceutical giants, like Merck, Pfizer, Amgen, and Biogen Idec to jump on the biosimilars bandwagon, many placing a focus on emerging markets in Asia and South America. Companies other than big pharma are also showing an interest in biosimilars. Many generic companies such as Teva, Hospira, Ratiopharm, Actavis and even non-pharma e.g. electronics giant, Samsung, are entering this new pharmaceutical market. Meanwhile, Novartis’ Sandoz unit has already proved itself to be a major player in the biosimilars market, being the only manufacturer to have three biosimilars on the market.



