

Pharmacovigilance at QPS
In today’s complex regulatory environment, even a single oversight in safety reporting can jeopardize an entire clinical program. Patient safety and regulatory integrity are non-negotiable. At QPS, compliance is the cornerstone of our pharmacovigilance services. Our team brings together expert leadership, global reach, and advanced technology to safeguard patients and support sponsors throughout every stage of clinical development.
Our validated systems are built to identify, document, report, and analyze safety data in full alignment with global regulations. By ensuring timely detection and evaluation of safety signals, we help sponsors remain audit-ready, mitigate compliance risks, and protect patients.
 Through continuous monitoring, proactive risk assessment, and seamless integration into your trial workflow, QPS delivers safety management that is reliable, compliant, and audit-ready. With QPS Pharmacovigilance, you can ensure participant safety, maintain regulatory compliance, and move forward with confidence.
Through continuous monitoring, proactive risk assessment, and seamless integration into your trial workflow, QPS delivers safety management that is reliable, compliant, and audit-ready. With QPS Pharmacovigilance, you can ensure participant safety, maintain regulatory compliance, and move forward with confidence.
At QPS, we have established robust systems to identify, record, report, and analyze safety data throughout a clinical trial. These systems are designed to ensure that any potential safety signals are quickly detected and thoroughly evaluated, allowing for rapid decision-making and intervention when necessary. Our proactive approach includes continuous monitoring and assessment, helping to mitigate risks and protect participant safety while maintaining compliance with regulatory requirements. By leveraging advanced tools and methodologies, we ensure that safety management is a seamless, integral part of the clinical trial process.
With QPS, you gain leadership-driven oversight, integrated medical monitoring, and future-ready technology, giving you the confidence to move your program forward safely and compliantly. Partner with QPS to make pharmacovigilance a seamless, reliable component of your clinical trial process. Let us help you deliver results with confidence and integrity.
Argus Safety Database
QPS has successfully implemented the Argus Safety Database, a premier pharmacovigilance system designed to support comprehensive safety case management for clinical trials. The adoption of the Argus Safety Database underscores QPS’s commitment to advancing patient safety, regulatory compliance, and operational excellence across its clients’ clinical drug development programs. Integrating this industry-standard safety platform strengthens QPS’ ability to capture, manage, and report adverse events in accordance with global regulatory requirements.
Why Sponsors Choose QPS Pharmacovigilance Services
Seasoned Leadership
Our leadership team provides expertise and oversight at every level:
- Safety Manager: 10+ years of pharmacovigilance experience across all clinical phases and post-market programs.
- Safety Officers: Positioned globally, delivering on-the-ground expertise and rapid responsiveness.
- Global Physician Reviewers: Apply real-world clinical experience for rapid, expert case evaluation.
- Executive Leadership Support: Strategic guidance, resource alignment, and program continuity.
Global Reliability
QPS delivers safety management that is reliable, compliant, and audit-ready.
- Worldwide Safety team with multiple layers of coverage.
- Continuous monitoring ensures compliance, data integrity, and uninterrupted service.
Powered by the Right Tools
We use the industry-standard Argus Global Safety Database for secure, scalable, and validated safety management.
- Secure & Scalable: QPS offers your own dedicated tenant hosted in our cloud-based, multi-tenant Argus database—ensuring data security, flexibility, and scalability across all clinical phases.
- Comprehensive Case Capture: Every SAE is logged directly in Argus, with streamlined intake via SAE notifications for accuracy and efficiency.
- Proven Workflows: Established processes clearly define project roles, track timelines, and keep your trial moving smoothly.
- Regulatory Confidence: Country-specific SUSAR reporting rules, powered by Regulatory Intelligence, ensure timely ICSR submissions and global compliance.
- Real-Time Insights: On-demand line listings provide instant access to safety data for faster, informed decision-making.
- Future-Ready: Argus scales seamlessly with your program, supporting you from early phase through post-market.
Safety Across All Stages of Your Trial
Planning
- Clear delegation of responsibilities and project-specific training.
- Comprehensive Safety Management Plan covering SAE reporting, medical review, follow-up, and regulatory submissions.
- Customized Argus setup tailored to your trial.
Implementation
- Ongoing Sponsor–Safety Manager communication.
- Regular database reconciliation for accuracy and compliance.
- Immediate notification of new case information.
- Sponsor review and approval of final case versions.
Closure
- Final database reconciliation to ensure data integrity.
- Delivery of submission-ready documents and audit-ready safety reports.
- Argus database scalability ensures continuity and readiness for your next study.
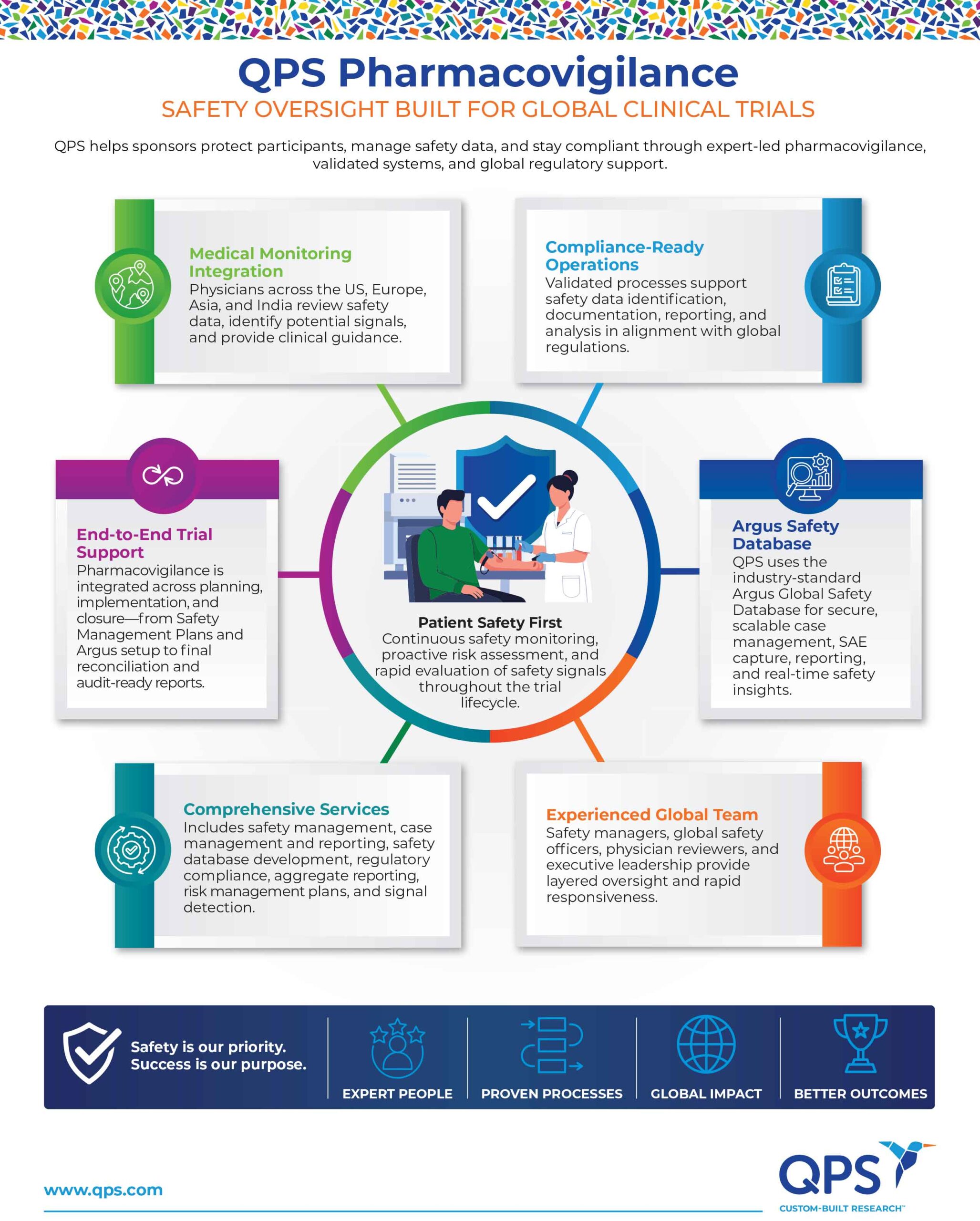
Our Pharmacovigilance Services
QPS delivers comprehensive pharmacovigilance support with clear, sponsor-focused deliverables designed to ensure patient safety and regulatory compliance at every stage of your trial, including:
Complete safety management forms the backbone of ensuring the safety of pharmaceutical products throughout their lifecycle, from development to post-marketing. Developing trial-specific Safety Management Plans (SMPs) and providing expert guidance to ensure regulatory compliance, structured oversight, and reliable safety operations is key. Effective safety management in pharmacovigilance focuses on the systematic collection, assessment, and mitigation of risks associated with the use of drugs and medical devices, with the ultimate goal of identify adverse events, assess safety signals, implement risk minimization strategies, ensure regulatory compliance, and protect patient health through timely and transparent communication.
Managing case intake for both medicinal products and devices, validating cases, entering data with MedDRA/WHO coding, and ensuring accurate expedited and periodic safety reporting through a clear query process. Comprehensive safety reporting in pharmacovigilance is a multi-faceted process that ensures the ongoing safety of pharmaceutical products. It encompasses the collection and analysis of adverse event data, timely reporting to regulatory agencies, signal detection, risk management, and continuous safety monitoring. By ensuring that safety data is accurately reported, analyzed, and acted upon, pharmacovigilance helps mitigate risks, protect patients, and ensure regulatory compliance.
Safety database development for pharmacovigilance refers to the creation and management of a specialized database designed to collect, store, and analyze safety data related to pharmaceutical products. These databases are crucial for monitoring the safety profile of drugs and medical devices throughout their lifecycle, ensuring patient safety, and meeting regulatory requirements. The safety database acts as the central repository for severe adverse events (SAEs).
At QPS, the preferred safety data base is Argus, as it is a premier pharmacovigilance system designed to support comprehensive safety case management for clinical trials.
Providing regulatory intelligence keep your program aligned with evolving FDA, EMA, ICH, and country-specific requirements, while delivering submission-ready documentation and integrating new technology standards. We also provide regulatory affairs services at QPS to support your client needs
Leveraging a secure Argus database with a dedicated tenant, continuous Safety Officer availability, and leadership oversight to deliver compliant, scalable solutions for every study.
Additional key Pharmacovigilance service include:
- Periodic Safety Update Reports (PBRER / PSUR )
- Development Safety Update Reports (DSUR)
- Annual Safety Reports (ASR)
- Line listings & summary tabulations
- RMPs prepared per ICH E2E, EU (GVP Module V), and local guidelines
- Initial RMPs and updates during lifecycle
- Product-specific safety concerns addressed with mitigation strategies Supports to build RMPs that are practical, compliant, and aligned with your product's global safety profile.
- Routine and ad hoc signal detection using validated methods
- Signal validation, prioritization, and assessment
- Expert ocumentation and regulatory communication (e.g., via RMP or PSUR)
Medical Monitoring
Our medical monitors partner with sponsors to:
- Review safety data to ensure clinical accuracy and relevance.
- Identify potential safety signals for early detection and intervention.
- Provide expert guidance on adverse events and participant safety.
By combining clinical insight with pharmacovigilance data, this service enhances trial safety oversight and supports end-to-end safety management across global programs. Through continuous monitoring and proactive risk assessment, we help sponsors maintain the highest standards of vigilance.
Aligned with FDA, EMA, and ICH guidelines, our medical monitoring services ensure that every case is managed with accuracy, accountability, and the confidence of expert clinical oversight. Together with pharmacovigilance, medical monitoring provides a unified approach to safeguarding patient safety and maintaining regulatory integrity across every stage of your trial.
Why Choose QPS?
- Proven Expertise: Decades of experience in pharmacovigilance and clinical trials with deep regulatory knowledge (FDA, EMA, MHRA, etc.)
- Advanced Technology: State-of-the-art tools for safety monitoring and reporting.
- Global Compliance: Adherence to international safety and regulatory standards.
- Participant-Centered Approach: Ensuring the safety and well-being of study participants.
Links to Relevant Documents
- Efficiently Performing Two Global Approval Studies in Prostate Cancer Patients
- Phase II-IV Clinical Overview
- Phase II-IV Clinical Services & Locations
- Early Phase Clinical Overview
- Expedited Study Delivery – Logistics
- Expedited Study Delivery – Pharmacy
- Studying Sedatives in Phase I Studies
- Pharmacokinetic Studies in Patients
- Corporate Overview Clinical Sites