

Market Situation
Prostate cancer is the second most frequent cancer diagnosis in men, and therapies to treat this disease are some of the most fiercely competitive in the oncology market.
Prostate cancer is the second most frequent malignancy (after lung cancer) in men worldwide, counting 1,276,106 new cases and causing 358,989 deaths (3.8% of all deaths caused by cancer in men) in 2018. The incidence and mortality of prostate cancer worldwide correlate with increasing age, with the average age at the time of diagnosis being 66 years. Although 2,293,818 new cases are estimated until 2040, a small variation in mortality will be observed (an increase of 1.05%). (Ferlay J EM, Lam F, Colombet M, Mery L, Pineros M, Znaor A, Soerjomataram I. et al. Global cancer observatory: cancer tomorrow. Lyon, France: International Agency for Research on Cancer)
Prostate cancer is also the fifth leading cause of death worldwide. Prostate cancer may be asymptomatic at the early stage and often has an indolent course that may require only active surveillance. Differences in the incidence rates worldwide reflect differences in the use of diagnostic testing. Prostate cancer incidence and mortality rates are strongly related to the age with the highest incidence being seen in elderly men > 65 years of age. (Prashanth Rawla. Epidemiology of Prostate Cancer. World J Oncol. 2019 Apr; 10(2): 63–89. PMID: 31068988, https://www.wjon.org/index.php/WJON/article/view/1191)
Age-adjusted incidence rates of prostate cancer have increased dramatically and this is largely because of the increased availability of screening for prostate-specific antigen (PSA) in men without symptoms of the disease. This PSA test leads to detection of many prostate cancers that are small and/or would otherwise remain undiagnosed, and which may or may not develop further into higher stage disease. (https://www.wcrf.org/cancer-trends/prostate-cancer-statistics/)
Download Case Study Here
QPS Involvement
A quickly growing biotech client approached QPS with a request to perform two Phase III trials in Prostate Cancer patients, designed to collect the efficacy, safety and pharmacokinetic information required to submit application for approval packages globally. The objective in mind was to conduct the studies and apply for global approvals in an expedited manner. QPS performed the duties of a central CRO, overseeing study conduct at all of the global clinical study sites across the USA, Europe and Asia.
To get those studies started QPS was also involved in the development process of new formulation for an existing marketed product indicated for treatment of prostate cancer.
Challenges
Due to the highly competitive nature of this market, the significant unmet global need for additional options in the therapeutic spectrum, and strict quality requirements for approval studies, the biotech client was keen to conduct the studies rigorously, but also as quickly as possible in US, Europe and Asia. That resulted in aggressive timelines and highly stringent expectations for exceptional quality in terms of project performance, administration and documentation as well as data across all global regions.
In addition, prostate cancer patients are, in general, elderly men, and they already have a variety of treatment choices available in most markets. This age group, coupled with existing available approved treatment options, made recruitment particularly challenging.
Solution
QPS focused on a three pronged approach to ensure that these trials were conducted to the highest possible standard and the results were delivered as quickly as possible: Recruitment, Project and Site Management as well as Data Quality.
Preclinical Research:
As the project was for the development of a group of three new formulations of a currently marketed drug indicated for treatment of prostate cancer, the development plan followed the 505(b)(2) regulatory pathway. Therefore, only limited toxicology studies in rodents were required to start clinical trials. Since this product was already a marketed product in a different formulation, determining ways to speed up the preclinical studies was a key step to accelerate the drug development cycle.
QPS Taiwan worked closely with the Sponsor to design, develop and support an efficient and tailored preclinical development program for the three different formulations. This program included the following studies:
- Toxicology studies: Dose Range Finding (DRF) and definitive studies f Bioanalysis: To analyze both the test article and biomarkers
- Formulation analysis
- Pharmocokinetic (PK) and Toxicokinetic (TK) analysis
- Quality studies: Tests to ensure the quality of drug product via Quality by Design (QbD) approach
As per standard practice at QPS Taiwan, our experts in Toxicology and Pathology assisted the Sponsor in responding the questions raised by the regulatory agency in a timely manner. With the step-wise strategy outlined above, the three different formulations moved forward to the clinical trial stage one-by-one.
Recruitment:
From the beginning of both trials, QPS Project Managers attached great importance to a close and well-maintained relationship with the study investigators. Working together with the sponsor, the study team did a great job establishing a strong global team spirit, rallying around the common cause of delivering another option for prostate cancer therapy to these elderly patients.
The studies began with well-organized investigator meetings (IMs) on all three continents, along with well-designed and delivered onboarding measures for all members of the study team. During these training meetings, all sites were fully informed and trained on the study protocols and procedures, and became motivated and committed to perform all study procedures to the highest possible standard. To ensure GCP compliance IM participants were trained on the study protocols and the latest GCP revisions.
To support the efficient alignment of the study teams in all three continents, the Global Project Management Team (GPMT) developed and delivered comprehensive regular Newsletter updates. These Newsletters included data on the current recruitment status at all sites and other relevant timely informational updates. In an effort to recruit study subjects as quickly as possible, recruitment between sites was designed to be competitive. Each Newsletter included a leaderboard, calling out and congratulating the best-performing sites and the highest performing region, based on recruitment progress to goal, as well as a series of other critical activities. This friendly competition and frequent updates on trending results likely contributed to the speedy recruitment and timely completion of the trial. In addition, the Newsletter functioned as a monthly reminder to the study sites that the GPMT was present and on point to provide comprehensive support to local site personnel.
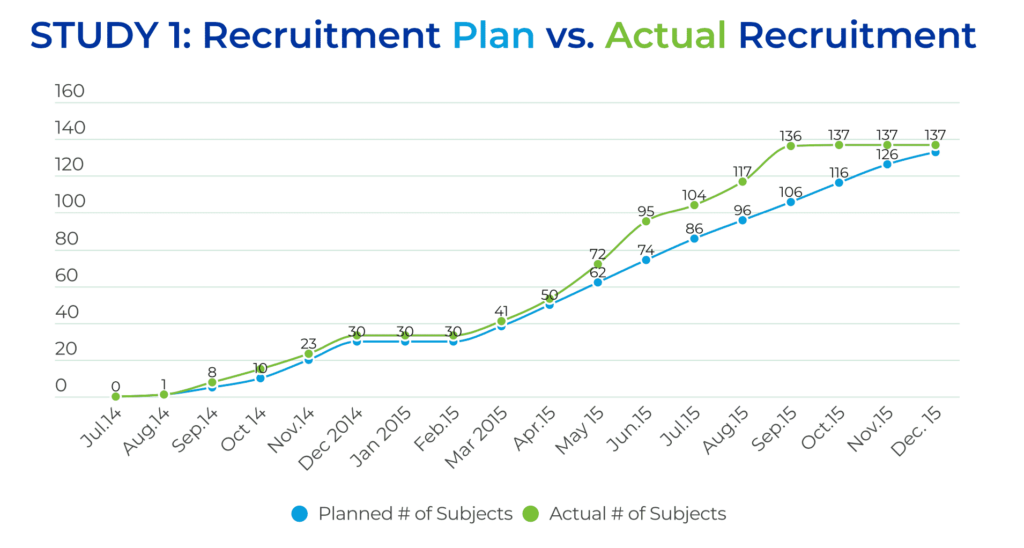
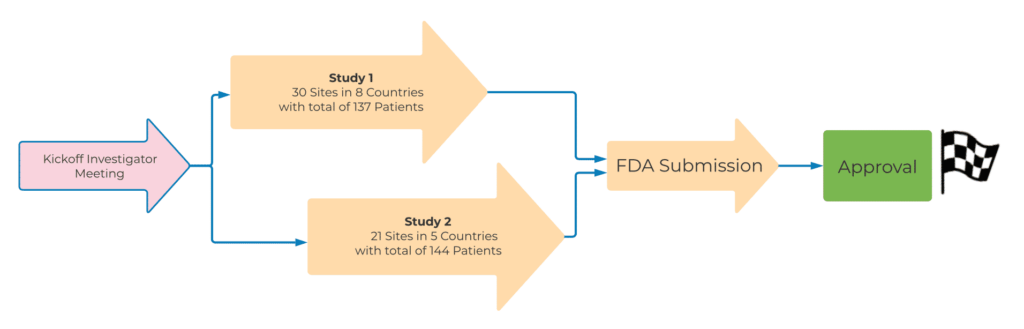
All of these efforts paid off during the study, resulting in excellent study recruitment results. Trial #1 consisted of two parts. Part I included enrolling 30 subjects, which triggered a recruitment pause of approximately 2.5 months for an interim analysis of the results. Part II planned to enroll an additional 103 patients. The overall recruitment goal for Trial #1 was to enroll 133 patients in 18 months. However, the QPS Global Clinical Operations team enrolled 137 patients, at 30 sites, across eight countries, in just 13.5 months, beating the enrollment goal by 4.5 months.
The US team enrolled approximately 47% of the patients at eight sites, and the EU team enrolled approximately 52% of the patients at 16 sites.
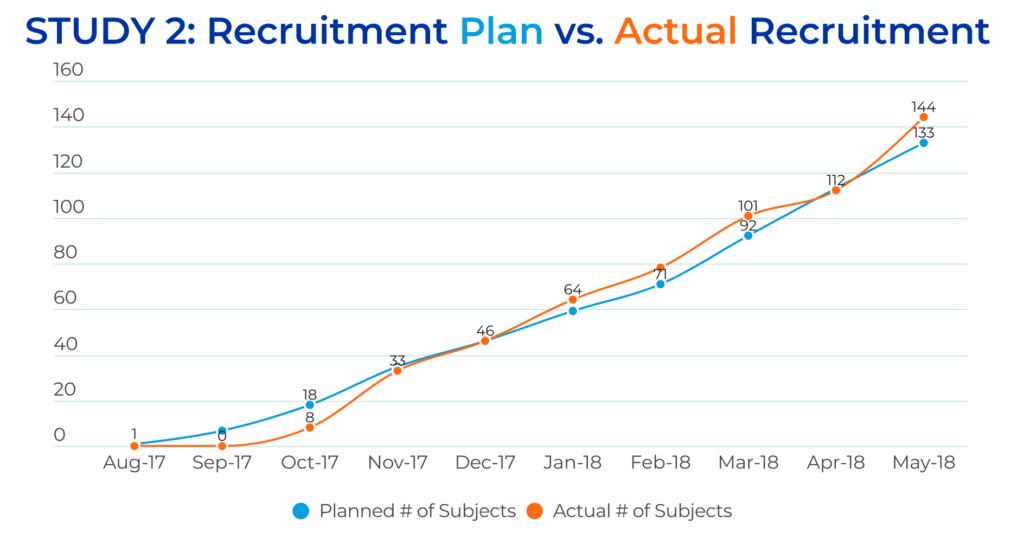
Based on the excellent results from Trial #1, ongoing close cooperation with investigators, an unchanged project and CRA team, and the addition of new sites, Trial #2 proceeded with the goal of recruiting approximately the same number of study subjects (n=133) in a significantly shorter enrollment period (ten months). The QPS Global Clinical Operations Team once again exceeded expectations, successfully enrolling 144 Prostate Cancer patients in just ten months.
Project Management:
Another important success factor was the seamless transition from Trial #1 to Trial #2 driven by a stable, experienced GPMT and CRA team. This highly experienced and scientifically educated team, based at QPS in Austria and across the EU, successfully used its central location within the different time zones to establish effective communication lines to all stakeholders across the USA, EU and Asia. From the beginning, the GPMT identified frictionless transfer of information to be a key success factor, especially in light of the tight timelines within this program. Both of these Pivotal Phase III trials benefited greatly from this seamless availability of all necessary information.
To further ease communication and streamline the process, the Sponsor and GPMT identified one investigator to lead and coordinate across both trials. This one primary investigator led the support of the Phase III program, up to and including the NDA application and approval process.
To ensure success, the GPMT thoroughly surveyed the past performances of all sites in advance, identifying the most effective sites and regions for prioritization. This process of prioritization enabled the team to focus on high-performing sites, ensuring they were appropriately motivated to secure high enrollment results.
Data Quality:
The GPMT, along with the Clinical Research Associates (CRAs), provided all necessary support to the study sites in order to complete trial-related activities, including data entry into the eCRF, in a timely and efficient manner.
The continuity, experience and scientific background of the local CRAs increased site performance resulting in outstanding data quality.
Finally, the GPMT, together with all stakeholders involved, attached great importance on the timely and correct set-up of all Trial Master File (TMF) elements, including data integrity and all other audit relevant aspects of both Phase III clinical trials. As a result, none of the sites’ TMF audits revealed any significant findings. In fact, all of the audits underlined the immaculate performance of these sites while conducting these critical Phase III studies, that were designed for submission to regulatory authorities to secure drug approval.
Regulatory:
QPS Biostatistics and Medical Writing teams supported the Sponsor with the fulfillment of regulatory requirements, such as preparing the Clinical Data Interchange Standards Consortium package, preparing the Bioresearch Monitoring inspection package, responding to FDA’s Biostatistics Information Request and revising the FDA Prescribing Information (PI). QPS supported the sponsor to help resolve issues and provide responses to meet FDA’s requirements in an accurate and timely manner after receiving FDA’s notification. With regards to the PI revisions, the QPS team assisted to confirm the FDA reviewers’ revisions and to clarify those in the package insert. This work included verification of FDA’s analysis, and communication with FDA to better understand their point of view and form a consensus. The QPS team also provided the Sponsor with an additional statistical analysis and an extra SAS dataset to meet the regulatory expectations efficiently and with good performance.
Results
QPS delivered two global Pivotal Phase III studies in prostate cancer patients, on time and with great data accuracy, enabling the Sponsor to complete and submit the application for marketing approval packages in an accurate and timely manner.
In completing these trials, QPS consistently outperformed recruitment targets, provided high-quality data, and delivered a flawless audit track. Keys to this performance included a strong operational team, diligent and timely training for all sites, robust support for principal investigators (PIs), and prompt data entry.
The overall result is that QPS provided significant support to create and assemble the data required to develop the regulatory application documentation for this new Prostate Cancer therapy. We are happy to report that this new treatment option has since been FDA approved.