


Introduction
Drugs bind to plasma proteins including albumin, α1-acid glycoprotein, and lipoproteins with different affinities and selectivities depending on the physical/chemical properties of the drug. It is generally accepted that the bound and unbound or free drug in plasma is in rapid equilibrium with each other, but it is the free drug that is biologically active, i.e., determines the biological activity of the drug. Therefore, it is important to determine the free fraction of drugs at different stages of drug development including discovery, preclinical, and clinical. At the discovery stage, plasma protein binding studies can be used to differentiate structural leads with different extent of binding. At the preclinical stage, the free concentration in different species can be used to estimate exposure multiples using data generated in preclinical efficacy models, GLP-compliant toxicology studies, and anticipated clinical dose range.
Factors that can alter the amount of free drug in plasma may include the concentration of the drug, concentration of the metabolite(s), the quantity and quality of the plasma proteins (e.g., malnutrition, infection, liver disease, renal disease, malignancy), and co-administration of two or more drugs. If a compound is highly bound to plasma proteins, it is important to determine which plasma protein the compound is bound to, e.g., albumin, α1-acid glycoprotein, lipoproteins, or other carrier proteins found in plasma. If the compound is highly bound to lipoproteins, it may be necessary to determine, using density gradient ultracentrifugation, which lipoprotein fraction, e.g., high density lipoprotein (HDL), low density lipoprotein (LDL), very low density lipoprotein (VLDL), etc. is involved.
Plasma protein binding drug-drug interactions (DDIs) can change the safety and efficacy profiles of a drug. This is especially clinically relevant for drugs with a narrow therapeutic index that are highly bound to plasma proteins, in which small changes in the free fraction can result in unexpected and often undesirable side effects. For example, if a drug has a therapeutic index of 5 and a free fraction of 1% (i.e., 99% bound), increasing the free fraction to 2% (i.e. 98% bound) could lower the therapeutic index to 2.5 due to a doubling of the free fraction.
Experimental Conditions
PROTEIN BINDING METHODS
Plasma protein binding determinations can be performed in vitro, ex vivo, or in vivo with radioactive or non-radioactive compounds using various methods including ultrafiltration, equilibrium dialysis, and microdialysis.
The choice of ultrafiltration, equilibrium dialysis or microdialysis depends on the type of data to be generated, in vitro, ex vivo or in vivo, and the physical/chemical properties of the compound being evaluated. A compound that binds non-specifically to equipment would dictate that equilibrium dialysis is used. Conversely, if the compound is unstable in plasma, ultrafiltration is the method of choice. However, if in vivo free concentration is the desired outcome, then microdialysis is the only option.
METHODS OF QUANTIFICATION
A robust, sensitive, and specific bioanalytical method, usually LC-MS/MS, or scintillation counting using a radioactive ligand, is required to quantify low, free concentration of the study drug.
OTHER FACTORS TO CONSIDER
When scintillation counting is chosen as the method of quantification, a radiolabeled test article can be spiked into the plasma samples collected. The amount of radiolabeled material added should be trace amount that does not change the binding characteristics. The purity of the radiolabeled material should be sufficiently high as not to affect the testing results.
Generating reliable plasma protein binding data requires: assessment of the compound’s stability during the duration of the protein binding study, amount of time required to reach protein binding equilibrium, and the extent of nonspecific binding of the compound to the equipment.
Using equilibrium dialysis, the extent of plasma protein binding of the test article is usually determined at 37° C for 4 – 6 hours in an apparatus that rotates to maximum interactions between the test article and the plasma protein binding sites.
Plasma samples can be obtained from preclinical studies, or from phase I, phase II, or phase III clinical studies conducted in healthy subjects or patients. The plasma may contain drug alone, drug with metabolites or drug and metabolites with co-administered drug(s) and its metabolite(s). The number of samples per subject or patient will depend on the study protocol’s specifics on number of subjects per dose, number of samples collected per subject and volume of blood collected at each collection time point.
Although the study design for a protein binding study is relatively straightforward for most compounds, the exact experimental conditions can be a challenge when there are specific questions being asked. The protein binding studies described in this White Paper show how ex vivo binding studies can answer some important clinical questions.
EXAMPLE 1
Question: What is the free fraction of Compound A in plasma of healthy subjects after a single dose?
Method: Plasma samples were obtained from a single dose escalation study in healthy subjects. Plasma samples were collected at 3, 4, and 6 hours post dosing from subjects who received the same dose. Pilot studies indicated that Compound A was stable in plasma at 37° C and was not bound to the ultrafiltration device. The ex vivo protein binding study was conducted at 37 C using ultrafiltration and the test article was quantified using LC-MS/MS.
Results:
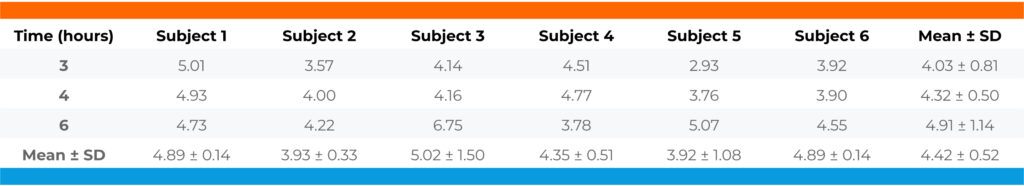
Table 1. The free fraction of Compound A in plasma of healthy subjects.
Conclusion: Compound A was highly bound to plasma proteins of healthy subjects with a free action averaging 4.42%. The percent free was independent of the time when the plasma sample was collected (p=0.345), suggesting it is independent of the plasma concentration of the test article and the concentration of the metabolite(s), if present.
EXAMPLE 2
Question: Is the free fraction of Compound B in plasma of healthy subjects after a single dose different from the free fraction after multiple doses?
Method: Plasma samples were obtained from a multiple dose escalation study in healthy subjects. Blood samples were collected at 1 hour post dosing from the same subjects on Study Day 1 and Study Day 9. Pilot studies indicated that Compound B was stable in plasma at 37° C and equilibrium was established after 3 hours of incubation. The ex vivo protein binding study was conducted at 37° C for 3 hours using equilibrium dialysis and the test article was quantified using LC-MS/MS.
Results:
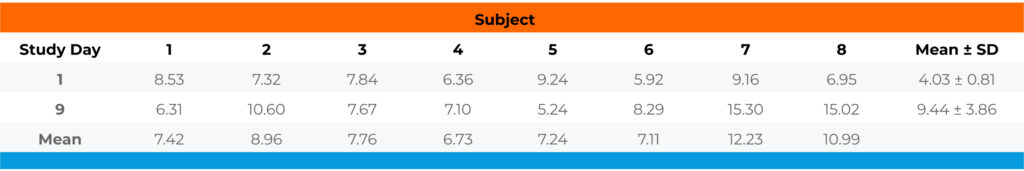
Table 2. The free fraction of Compound B in plasma of healthy subjects after single and multiple doses.
Conclusion: Compound B was moderately bound to plasma proteins of healthy subjects with a free fraction averaging 7.67% ± 1.25 on Study Day 1 and 9.44% ± 3.86 on Study Day 9. There was no significant difference in free fraction between Study Day 1 and Study Day 9 (p=0.256).
EXAMPLE 3
Question: Does renal impairment affect the free fraction of Compound C?
Method: Plasma samples were obtained at 3 hours post dosing from a single dose study comparing the pharmacokinetics of Compound C in healthy subjects, and renal impaired patients. The ex vivo protein binding study was conducted at 37° C using ultrafiltration, and the test article was quantified using LC-MS/MS.
Results:
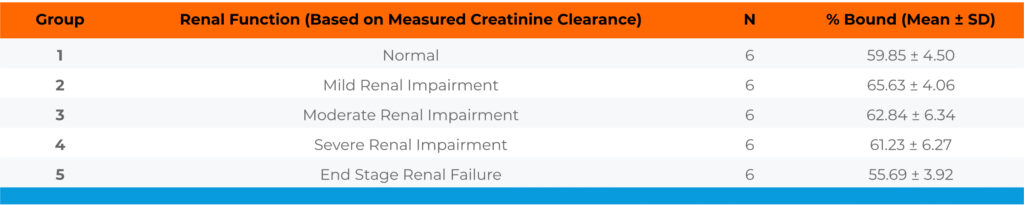
Table 3. Protein binding of Compound C in plasma of healthy subjects and renal impaired patients
Conclusion: Compound C was moderately bound to plasma proteins of healthy subjects with a free fraction averaging 40.15%. The free fraction of Compound C in patients with different levels of renal impairment was similar to the free fraction determined in healthy subjects.
EXAMPLE 4
Question: What is the free fraction of Compound D and its major metabolite X?
Method: Plasma samples were obtained at 2 and 4 hours post dosing from a singe dose, escalation study. The ex vivo protein binding study was conducted at 37° C using equilibrium dialysis, and the test article was quantified using LC-MS/MS.
Results:

Table 4. The free fraction of Compound D and its major metabolite X in plasma of healthy subjects.
Conclusion: Compound D and its metabolite X were tightly bound to plasma proteins of healthy subjects with a free fraction averaging 0.213% ± 0.053 and 0.243% ± 0.059, respectively. The free fraction of Compound D and metabolite X appeared to be independent of their plasma concentrations over a 33-fold and 49-fold range, respectively.
EXAMPLE 5
Question: Does Compound E affect the free fraction of Compound F?
Method: Compound F was administered to healthy subjects once daily for 19 days, and Compound E was co-administered with Compound F three times daily on Study Days 15 to 19. Plasma samples were obtained at various time points on Study Day 14 (steady state plasma concentrations of Compound F) and Study Day 19 (steady state plasma concentrations of Compound E and Compound F). The ex vivo protein binding study was conducted at 37° C using equilibrium dialysis, and the Compound F was quantified using LC-MS/MS.
Results:

Table 5. The free fraction of Compound F in the absence and presence of Compound E in plasma of healthy subjects.
Conclusion: Compound F was highly bound to plasma proteins of healthy subjects with a free fraction averaging less than 1%. The free fraction of Compound F, as well as the free concentrations of Compound F, were not changed in the presence of Compound E. These results indicate the lack of protein binding interactions after co-administration of Compound E and Compound F.
SUMMARY
The ex vivo protein binding studies presented in this White Paper show the type of data that can be generated, and the value of knowing the extent of plasma protein binding in guiding the design and execution of the clinical development plan for a drug.
