


The 505(b)(2) Pathway
Today, many pharmaceutical companies – generic or innovative, large or small, privately held or publicly traded – are choosing the 505(b)(2) New Drug Approval (NDA) regulatory pathway. The 505(b)(2) NDA route relies on investigations not conducted by or for the applicant, or on investigations for which the applicant has not obtained a right of reference or use from the person who conducted the original investigations1. To submit 505(b)(2) NDAs, applicants rely on published literature of clinical studies and/or the FDA’s filing of safety and efficacy data for a previously approved drug. Many pharmaceutical companies are unaware of the resources required to maximize the benefits of 505(b)(2) applications.
THE HATCH-WAXMAN ACT
Amendments made to the Federal Food, Drug, and Cosmetic Act (FDCA) in 1984 were incorporated into the Drug Price Competition and Patent Term Restoration Act, commonly known as the Hatch-Waxman Act. This Act was created to find common ground for promoting innovation and allowing generic competition in the pharmaceutical industry. What the Hatch-Waxman Act does is provide an expedited means for obtaining approval for a generic drug through an abbreviated new drug application (ANDA) that builds upon the innovating company-sourced proprietary safety and effectiveness data of a drug that has already received Food and Drug Administration (FDA) approval2.
THE 505(B)(2) NDA OPTION
The 505(b)(2) NDA option was initiated through the Hatch-Waxman Act. A 505(b)(2) application is slightly different from an ANDA. An ANDA proves that the product is identical to a previously approved product based on chemistry and bioequivalence data, without the need for preclinical or clinical data for safety and efficacy. A 505(b)(2) NDA, on the other hand, requires a combination of new preclinical or clinical data and already approved studies. Moreover, 505(b)(2) applications qualify for 3 or 5 years of market exclusivity, whereas ANDAs only qualify for 180 days.
You can determine whether your drug is suitable for approval via the 505(b)(2) NDA route by checking the FDA’s 1999 draft guidance Applications Covered by Section 505(b)(2). According to the guide, applications for which a pharmaceutical product may be approved via the 505(b)(2) route include, but are not limited to, the following:
- Change of dosage form or dosage regimen
- Conversion to lower or higher strength
- Change in the route of administration
- Substitution of an active pharmaceutical ingredient in a combination product
- Change in formulation
- New molecular entity (NME)
- Conversion from prescription drug (Rx) to over-the- counter (OTC) drug
- Drug with new ingredients from animal or botanical sources (naturally derived or recombinant)
- A bioequivalent product
SUMMARY OF BENEFITS
Benefits of 505(b)(2) NDAs include:
- Relying on already established and approved safety and efficacy data (only limited new clinical data required)
- Patent protection for any modifications
- 3 or 5 years market exclusivity: 5 years of exclusivity for the first approval of a new chemical entity, and 3 years of exclusivity after an active pharmaceutical ingredient (API) has been approved for the first time, in a drug requiring one or more new clinical studies. If product is for an orphan disease, exclusivity could be up to 7 years.
NDA APPROVAL STATISTICS
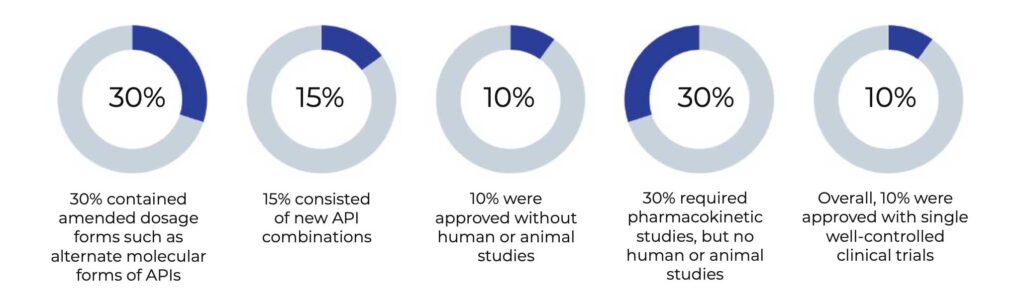
At least 185 505(b)(2) NDAs have been approved since the Hatch-Waxman amendments. Out of 115 approvals between 1998 and 2009, 30 percent contained amended dosage forms such as alternate molecular forms of APIs, and 15 percent consisted of new API combinations3. Ten percent were approved without human or animal studies. An additional 30 percent required pharmacokinetic studies, but no human or animal studies. Overall, 10 percent were approved with single well-controlled clinical trials3.
Out of 115 approvals between 1998 and 2019
IDEAL FOR SMALLER PHARMACEUTICAL FIRMS
Many pharmaceutical companies, generic as well as innovative, are leveraging their resources and making a shift toward the potential added value and profit associated with complex drug formulations. Smaller pharmaceutical firms do not usually have the in-house resources for research to file an NDA for a new chemical entity. Those firms are also unlikely to have the full in- house capacity needed to do the work of filing or to conduct a thorough literature review to investigate earlier studies on reference drugs, and often benefit from hiring external experts or consultants with a wealth of experience in fulfilling the complex requirements of 505(b)(2) NDA applications to get the job done. Contracting integrated 505(b)(2) NDA services is more cost and time-effective for such firms3. Such services can cut costs and shorten timelines by avoiding repetition of clinical trials of previously approved drugs.
WHY QPS SHOULD BE YOUR CHOICE
Obtaining approval via the 505(b)(2) NDA route is a great idea. Choosing QPS as your expert consultant is an even greater idea. QPS is a global leader in contract research. We have highly experienced staff, top-notch resources, and the in-house capacity to fulfill your needs.
QUALITY PHASE I STUDIES
Our service is recognized as one of the top in the industry for complex, high quality early stage (Phase I) clinical research. QPS provides fully integrated Phase I services, including protocol development, clinical protocol advice, clinical trial conduct, bioanalysis, and data management/ statistical analysis. QPS can also conduct clinical studies in various patient populations to support your application for marketing approval.
STATE-OF-THE-ART PHASE I SITES
QPS provides access to state-of-the-art Phase I sites around the globe. These sites offer extensive databases of healthy volunteers in rich recruiting environments, and employ highly-skilled and experienced staff who use disciplined processes to deliver the highest accuracy and data integrity possible.
REFERENCES
1. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER).Guidance for Industry Applications Covered by Section 505(b)(2), Draft Guidance [Internet]. 1999. Available from: http://www.fda.gov/downloads/DrugsGuidances/ucm079345.pdf
2. Danzis SD. The Hatch-Waxman Act: History, Structure, and Legacy. Antitrust Law J. 2003 Jan 1;71(2):585–608.
3. Gelber L. Trends in 505(b)(2) Approvals [Internet]. Contract Pharma. [cited 2015 Feb 20]. Available from: http://www.contractpharma.com/issues/2010-03/ view_features/trends-in-505- b-2-approvals/
