


Bioequivalence And Bioavailability Trials Are Essential Components Of Generic Or Hybrid Medicinal Product Applications
Such studies are also conducted to support formulation selection and development. Deciding on the best site or geographical area in which to conduct the trial is key. While timelines and budgets are important, other factors must be taken into account, as will be explained in this paper. Although the focus here is on bioequivalence studies, most of the requirements and considerations discussed are applicable to other Phase I trials as well.
The first question is, where will the product be registered? This white paper focuses on trials that will support registration of a medicinal product in the EU. In such cases, regional authorities establish trial requirements as set forth in the marketing authorization application (MAA). For drug applications in the US, via abbreviated new drug applications – abbreviated new drug applications (ANDAs), the regulatory requirements are similar but based on a separate set of laws.
BASIC EU REQUIREMENTS
The three key essentials for clinical trials supporting registration in the EU are:
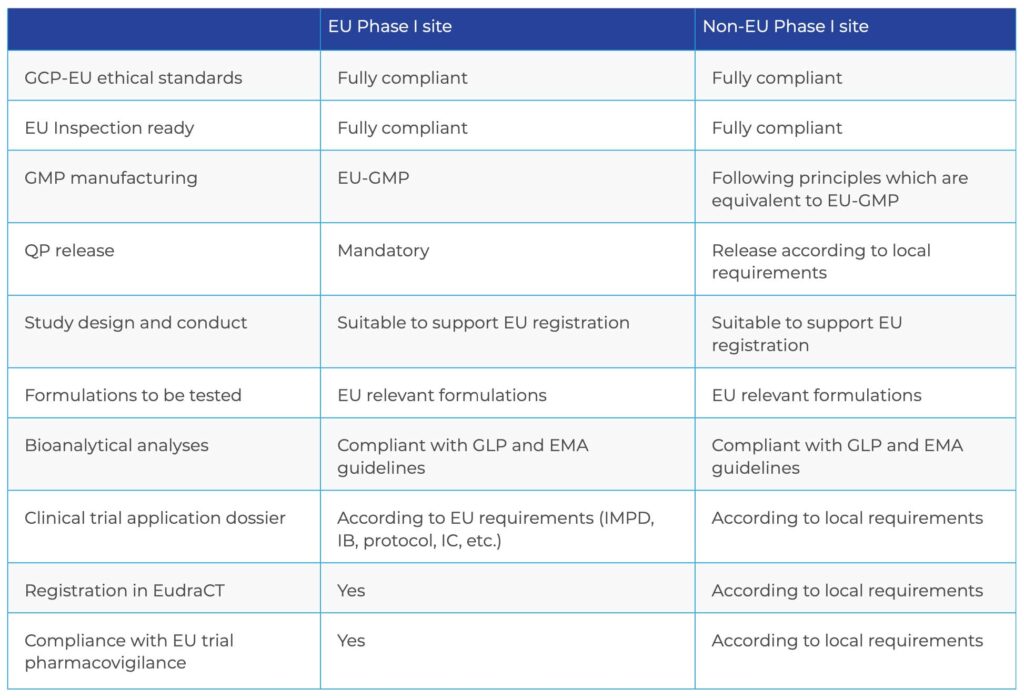
- The conduct of the trial complies with EU ethical standards and Good Clinical Practice (GCP).
- The trial design and conduct allow a valid conclusion to be drawn from the results, in line with the European Medicines Agency Committee for Medicinal Products for Human Use (EMA-CHMP) guidelines and other requirements for marketing authorization of medicinal products in the EU.
- The trial conduct complies with all local requirements.
As long as these three principal requirements are upheld, the investigational site may be located anywhere in the world. The following sections explain in more detail what meeting these conditions involves.
KEY REQUISITE #1: ETHICAL STANDARDS AND GCP REQUIREMENTS
- Clinical trials conducted in the EU must comply with GCP as specified in the EU Clinical Trials Directive 2001/20/EC, the GCP Directive 2005/28/EC and related guidances.
- Clinical trials conducted outside the EU, but included in an MAA for the EU market, must be conducted according to principles equivalent to those in the EU GCP Directive and must uphold ethical standards equivalent to those applied in the EU, including adherence to the Declaration of Helsinki. The EMA reflection paper (EMA/121340/2011) provides detailed information on this topic.
EU and non-EU trial sites can be inspected by EU GCP inspectors at the request of EU regulatory agencies. Trial sites must fully facilitate such inspections, providing access to all information requested by the inspectors and responding to questions or findings in a timely and constructive manner.
KEY REQUISITE #2: VALID TRIAL DESIGN AND CONDUCT
- To demonstrate bioequivalence, the trial design must comply with the EMA-CHMP Guideline on the Investigation of Bioequivalence (CPMP/EWP/ QWP/1401/98) and/or other applicable EMA-CHMP guidelines, e.g. guidelines for a specific type of formulation, unless otherwise justified.
- The test formulation used in the study should be representative of the product to be marketed in the EU.
- The reference formulation must be the selected EU reference medicinal product. Use of a reference formulation obtained from a local non-EU market is likely to be rejected by the authorities.
- The trial population must be suitable for investigating the absorption and other pharmacokinetic characteristics of the compound. In principle, the trial population may include any subjects, regardless of race, gender or other characteristics. In most cases, a homogeneous adult male population is preferable. In some situations, the characteristics of the compound being studied restrict the trial population and may influence the choice of location.
- If the study must be conducted under fed conditions (e.g. intake of medication after a high fat breakfast), it is advisable that the volunteers are accustomed to such a food pattern. In these situations, speed of recruitment may vary according to location.
- The bioanalytical samples must be analyzed according to the EMA Guideline on Bioanalytical Method Validation1 and in accordance with Good Laboratory Practice (GLP). This guidance is a bit dated, and there is an updated draft guideline currently out for comment2. Although the bioanalytical lab working in compliance with these guidelines can be anywhere in the world, use of a local laboratory may be advantageous, for instance, when rapid interim results are needed.

KEY REQUISITE #3: LOCAL REQUIREMENTS
-
For trials conducted within the EU, certain requirements apply such as:
A trial application dossier in the EU format
Registration in the European Clinical Trials Database (EudraCT)
A designated Legal Representative located in the EU
Compliance with EU Clinical Trial Pharmacoviligance - For trials conducted at sites outside the EU, it is best to refer to local regulations and authorities or consult the site to ascertain local requirements.
CONSIDERATIONS FOR GMP AND IMPORTED MEDICATION
- For trials conducted within the EU, the principles of Good Manufacturing Practice (GMP) should be applied to all investigational medicinal products used in the study (Directive 2003/94/EC). If the medication is manufactured outside the EU, the clinical trial batch must be released by an EU Qualified Person (QP).
- Medication used in trials in non-EU countries must be manufactured following principles equivalent to EU- GMP. For pivotal bioequivalence studies, adherence to EU-GMP for the test formulation will facilitate the marketing authorization (MA) approval.
- If the investigator’s site and the pharmaceutical formulation manufacturing site are not in the same geographical area, customs clearance of the trial medication needs to be arranged. This is often only initiated once the trial has been approved, and thus may affect the overall trial timeline.
SUMMARY AND CONCLUSION
For MA applications, EU regulatory agencies accept studies conducted in non-EU countries if the studies meet compliance criteria equivalent to those required of trials conducted in the EU, and the study design supports EU product registration.
Selecting the location of a clinical Phase I trial site is a case-by-case decision, and pros and cons vary for different situations.
There are 3 Key Requisites for consideration in this decision: Ethical Standards and GCP Requirements, Valid Trial Design and Conduct, and Local Requirements. Meeting and exceeding those considerations is critical for seamless movement past Phase 1 and towards EU regulatory approval.
COMPARISON OF REQUIREMENTS FOR A PIVOTAL BIOEQUIVALENCE STUDY WITH A FORMULATION INTENDED TO BE MARKETED IN THE EU
REFERENCES
1. EMA Guideline on Bioanalytical Method Validation (https://www.ema.europa.eu/en/bioanalytical-method-validation).
2. EMA Guideline on Bioanalytical Method Validation Draft for comment: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation
