


Introduction
Development of DNA or RNA analogs that can block the activity of selected genetic sequences makes the design of highly specific therapeutic treatments for various diseases possible. Therapeutic oligonucleotides, or more specifically, siRNAs are being developed for treatment of hepatitis C virus, age-related macular degeneration, cholesterol control and even cancer therapies. The effect of small strands of siRNAs on gene expression has had a tremendous impact on basic and applied pharmaceutical research. siRNA gene silencing is currently one of the most promising and fastest growing new approaches for disease therapy.
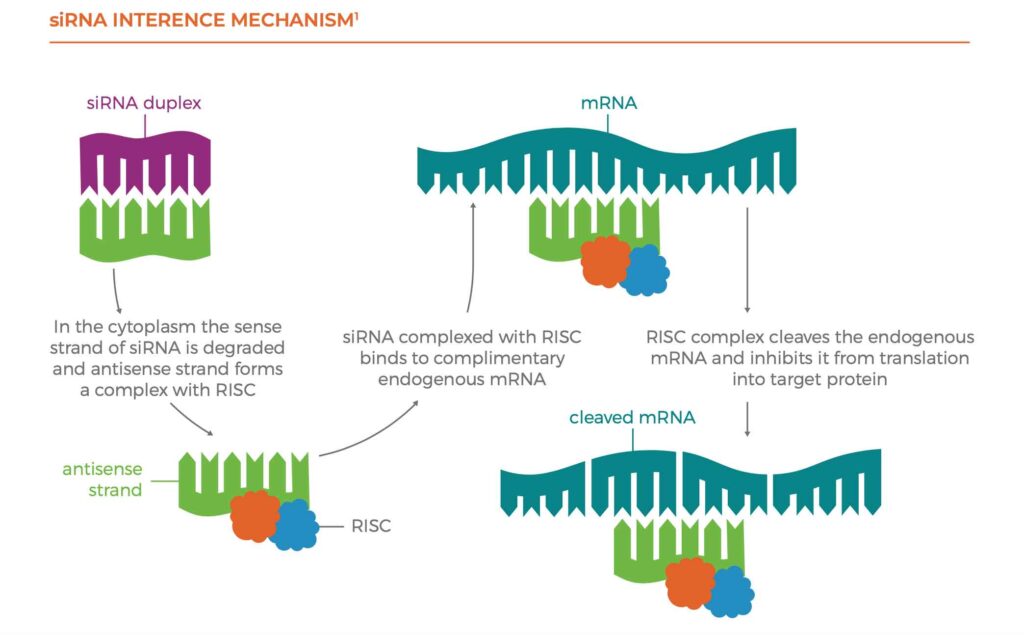
The siRNA interference mechanism is given below. First, the siRNA duplex is transported across cellular membranes into the cytoplasm with the help of the sense strand. The antisense strand of the siRNA is incorporated into the RNA- induced silencing complex (RISC), where the antisense and sense strands are separated. The RISC containing the antisense strand seeks out and binds to complementary mRNA sequences. These mRNA sequences are then cleaved by an enzyme within the RISC responsible for mRNA degradation, which leads to mRNA down-modulation of the target protein.
BARRIERS ENCOUNTERED WITH DELIVERY OF SIRNA THERAPIES
Several barriers exist that limit the use siRNAs in effective and controlled gene silencing. The main issue is transporting the drug into the cytoplasm of the target cell. A siRNA complex must travel through the blood stream, avoiding enzymatic degradation by nucleases found in the blood and kidney. Metabolism by 3’ exonuclease enzyme, so named since it attacks the 3’ terminal end of the siRNA oligonucleotide, is the major nuclease active in plasma. However, cleavage of bonds at the 5’ terminus and even the inner phosphate linkages (by endonucleases) can also take place. Filtration by the kidney also plays an important role in siRNA clearance.
Free siRNA is not readily taken up by cell cytoplasm, the hydrophobicity and negative charge of siRNA molecules prevents them from easily crossing biological membranes. This is addressed by the use of guide or sense strand RNA that has structural features that improve transport of the antisense/sense duplex across cellular membranes and bind with high affinity and specificity to the target site.
These barriers are overcome by modifications of the structural architecture of the drug. These modifications make the siRNA more nuclease-resistant by minimizing degradation and enhancing the overall stability of the siRNA. These structural modifications are also designed to improve circulation time and tissue uptake of the drug. The performance of siRNA therapeutics are improved by chemical modification to sites along the siRNA strand including the ribose sugar, the base, phosphorous acid linkages and end groups of both sense and antisense strands. A common solution is the replacement of the phosphodiester (PO4) group with phosphorothioate (PSO3) which inhibits enzymatic degradation by reducing siRNA’s susceptibility to exonucleases. The introduction of an O-alkyl group or a fluorine group at the 2’ position of the ribose sugar also results in longer half-lives and increased RNA interference activities. Development of several other modifications on end groups of the sense strand serve to reduce hydrophobicity of the duplex, which improves transport across cellular membranes. The basic goal of a successful modification is enhancement of siRNA serum stability without negative effects on its gene silencing activity.
THE ROLE OF METABOLITE IDENTIFICATION AND QUANTITATION IN SIRNA DRUG DEVELOPMENT
Better understanding of metabolites and metabolic pathways can influence strategies that improve drug efficacy and safety. Since siRNAs are non-natural molecules, the degradation/metabolism of modified siRNAs demands thorough monitoring of new metabolites formed, as there is a potential for toxicity with unstudied metabolites. Advances in analytical technology, particularly LC/HRMS, have enhanced capabilities to generate metabolite and pharmacokinetic data with lower detection limits and much more specificity. Even so, some metabolites evade detection due to poor ionization or large differences in detector response. Historically, the approach in this case is to utilize a metabolite standard for both structural confirmation and direct quantitation when synthetic standards of metabolites are available.
The method of choice for generating comprehensive metabolite profiles without standards is radio- chromatography. If a test compound cannot be effectively radiolabeled or radiolabel is lost due to metabolism, an authentic standard for the metabolite is needed for absolute quantitation. If no synthetic standards are available, some steps can be taken to measure metabolites in alternate ways to determine or eliminate the need for more toxicology studies. Generally, the probability of having to perform an additional in vivo toxicology study with an unexpected metabolite is kept quite low by designing experiments that assure representative coverage of all metabolites above 10% of dose. Performing a quantitative coverage assessment for a matrix is accomplished by mixing equal volumes of appropriately pooled samples. This, followed by an analysis using full-scan HRMS, demonstrates full metabolite coverage.
PROCEDURES FOR A TYPICAL SIRNA METABOLITE PROFILING AND QUANTITATION STUDY
The procedures described here outline fit-for-purpose activities to understand more clearly parent and metabolite coverage in an siRNA metabolism study. The required sample-pooling scheme represents the time course fate of the drug over the entire experiment. Once pooled, an extraction step is performed on each sample to concentrate the oligonucleotides and clean up interfering compounds in the matrix. Finally, all samples are qualitatively and quantitatively, or semi-quantitatively, analyzed by LC/HRMS.
Sample pooling – If mass balance is required, a pooling scheme must be developed. For radiolabeled studies, the total radioactivity determines the pooling scheme. In cold studies, plasma, bile or urine samples are pooled by output volume across all animals by time for quantitation and a single sample pooled across all time points is used for metabolite profiling. Fecal samples are pooled by equal percent weight across animals at each time interval. Tissue samples such as liver, kidney, muscle etc. are processed similarly to feces but they are typically not pooled.
Extraction – After sample pooling, an extraction step, usually liquid-liquid or stationary phase extraction (SPE) is designed to concentrate analytes and metabolites and to clean up the sample matrix. There can be large differences between a liquid-liquid extraction and SPE extraction and if there has been no prior method developed for the analyte, both approaches should be explored. For an acceptable mass balance study, it is critical for extraction recovery to be determined for each matrix and a predetermined allowable recovery achieved.
LC/HRMS Analysis – Finally, all samples are qualitatively and quantitatively analyzed by ion pairing-LC/neg ESI-HRMS (IP-LC/HRMS). The high-resolution capabilities of the mass spectrometer allows for quantitative analyses with short LC gradients. Often chromatographic peak overlap requires a longer, shallower liquid chromatographic gradient for the best metabolite identification.
Metabolite Identification – Metabolite identification is performed by obtaining full scan total ion chromatogram data for all profiling samples. Potential metabolites are identified by comparison to control or zero time-point data. This can be done manually or using state-of-the- art oligonucleotide metabolite search software which uses accurate mass, charge state (multiple), isotopic distribution and intensity to locate and propose an oligonucleotide identity. Due to the potentially high number of metabolites that may be present, only those with significant intensity and good quality mass spectra are reported. However lower level metabolites are often useful to explain biotransformation pathways.
Quantitation – Initially, a quantitation method using parent drug and any synthesized metabolite standards is developed. Closely related internal standards are used to correct for any sample preparation and sample introduction deviations. Amounts of unexpected metabolites are estimated by simple comparison of spectral area counts to parent or a related metabolite. For this, all mass spectral responses are assumed to be equivalent across components.
THE FUTURE OF THERAPEUTIC OLIGONUCLEOTIDE METABOLITE PROFILING AND QUANTITATION
Therapeutic oligonucleotides targeting new diseases, such as cancer, viral infections, autoimmune diseases, and neurodegenerative diseases are being developed. Improved drug delivery methods, such as mRNAs, cyclic RNAs, RNA micelles and many others, all create new analytical challenges. As broader approaches evolve, current analytical methods for oligonucleotides and associated metabolites must evolve with them. So, new bioanalytical techniques will become indispensable to comply with new regulatory requirements.
There is a need for separation science improvements; stationary phase capacity, selectivity for these large molecules and more efficient ion pairing liquid chromatography. New developments in microflow chromatography will have more and more of an effect on sensitivity for these compounds. There is a need for improved mass spectrometer detector sensitivity, new ionization technology and mass analyzer design. These technological advances have the potential to revolutionize therapeutic oligonucleotide analysis.
BENEFIT FROM THE WORLDWIDE RESOURCES THAT A GLOBAL CONTRACT RESEARCH ORGANIZATION BRINGS
Whether your focus is small molecules, protein biotherapeutics, vaccines, gene therapy or cell therapy, QPS provides a full range of bioanalytical services to support all drug development needs from discovery, through clinical development and regulatory filing.
REFERENCE
1. Huntington Disease Research Group. Homepage, University of Toronto, Canada (2010), http://individual.utoronto.ca/rpersaud/#.
