


Executive Summary
Biosimilars have advanced from a promising idea to a proven pathway for expanding patient access to biologic therapies and easing pressure on healthcare budgets. Today, sponsors that succeed in biosimilars do two things well: they build a credible similarity story and support it with regulator-ready bioanalytical evidence.
Regulators have matured their expectations. The most successful programs use a stepwise approach in which analytical characterization and comparative bioanalysis reduce uncertainty early—often minimizing the need for extensive latestage clinical efficacy programs.
This paper provides a snapshot of how biosimilars developed, where the market is heading, what regulators expect
today and how QPS helps sponsors deliver the data packages that accelerate global approvals.
What Is A Biosimilar—And Why It Is Not A Generic Biologic
A biosimilar is a biologic medicine that is highly similar to an already approved reference biologic and has no clinically meaningful differences in safety, purity and potency. Because biologics are produced in living systems, exact replication is not feasible. Biosimilars must demonstrate similarity through a scientific comparability exercise rather than chemical identity.
The European Medicines Agency (EMA) emphasizes that biosimilars are not regarded as generics because manufacturing complexity and natural variability prevent exact replication at the molecular micro-heterogeneity level.1
For developers, this means biosimilars succeed when the development strategy produces a defensible endto end similarity narrative—anchored by analytical characterization and strengthened by robust comparative bioanalysis.
A Brief History: How Biosimilars Earned Regulatory Confidence
Europe approved the first biosimilar in 2006, establishing a centralized pathway that helped define what “highly similar” means in practice. The United States followed with the Biologics Price Competition and Innovation Act (BPCIA) of 2009, creating an abbreviated approval pathway for biosimilars and interchangeable biologics.2
Over time, biosimilars have expanded beyond early protein classes to include high-impact monoclonal antibodies and other complex therapies. Regulatory experience has steadily increased, and global guidance documents now provide clearer direction on analytical, pharmacokinetic (PK), pharmacodynamic (PD) and immunogenicity expectations.
The 2026 Approval Landscape: Scale, Maturity and Competitive Density
As shown in Figure 1, approvals of biosimilar drugs in the US and EU have increased steadily over the last five years. In the US, 82 biosimilars had been approved as of January 2026.3 In Europe, more than 140 biosimilars have been authorized or recommended through the EMA pathway, reflecting Europe’s long-standing leadership in this category.4
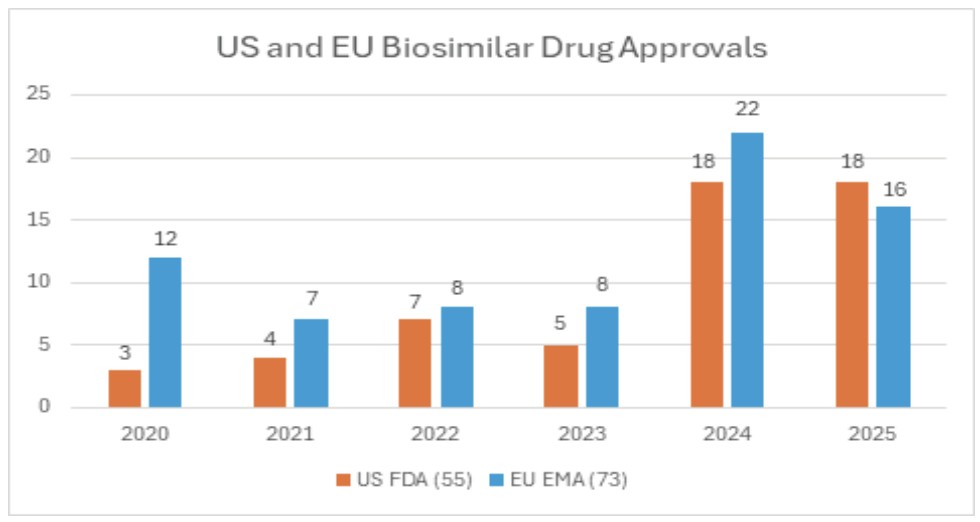
Figure 1. Biosimilar drug approvals in the US and EU, 2020-2025.
Therapeutic Areas and Reference Products
Biosimilar competition is concentrated around major biologic reference products in:
- Oncology (e.g., trastuzumab, bevacizumab and rituximab)
- Immunology and autoimmune disorders (e.g., adalimumab, infliximab and etanercept)
- Ophthalmology (e.g., ranibizumab and aflibercept)
- Endocrinology and diabetes (e.g., insulin products)
- Bone disease and supportive care (e.g., denosumab)
Monoclonal antibodies represent the largest segment of the biosimilars market by drug class, accounting for approximately 44.9 percent of revenue share in 2025.5
Market Momentum and Growth Outlook
Biosimilars began as a regulatory innovation intended to introduce competition into the biologics market. In 2026, they represent both a mature scientific pathway and a strategic growth opportunity. With greater regulatory clarity, market acceptance has expanded, and competitive intensity is rising.
The global biosimilars market was estimated at $39.59 billion in 2025 and is projected to reach $151.58 billion by 2033, representing a compound annual growth rate of 18.44 percent from 2026 to 2033.5
Key drivers include patent expirations of blockbuster biologics, increasing prevalence of chronic disease, payer pressure to reduce drug costs and growing confidence in regulatory pathways.5
The direction is clear: biosimilars are now a central component of biologics competition worldwide.
Why Biosimilars Matter: Access, Competition And System-Wide Savings
Expanding Patient Access
Biologics have transformed care in oncology, immunology, endocrinology, ophthalmology and rare diseases. However, high cost can limit access. Biosimilars introduce competition that broadens treatment options and reduces financial burden for patients and health systems.6
Demonstrated Economic Impact
In the US, biosimilars are delivering measurable savings. The Association for Accessible Medicines reported that biosimilars have contributed to $56.2 billion in healthcare savings since 2015, resulting in nearly 3.3 billion days of patient therapy over that period.6
These savings reflect not only reduced costs but also expanded access to biologic treatments for patients who might otherwise delay or forego therapy.
Today’s Regulatory Model: Stepwise Proof And Totality Of Evidence
Regulators in the US and EU apply a stepwise approach to biosimilar approval:
- Analytical similarity
- Nonclinical studies, as appropriate
- Clinical studies focused on pharmacokinetic (PK) and pharmacodynamic (PD) data
- Randomized, controlled trials to evaluate safety, efficacy and immunogenicity
The US Food and Drug Administration (FDA) evaluates biosimilars using a totality-of-evidence framework in which comparative analytical and clinical data are considered alongside the agency’s prior findings regarding the reference product.
Clinical programs are comparative and purpose-built to resolve residual uncertainty. Strong analytical and PK similarity data can significantly reduce the need for large confirmatory efficacy trials.
Interchangeability
Interchangeability is a distinct designation under US law. Sponsors must provide additional evidence demonstrating that the biosimilar can be expected to produce the same clinical result as the reference product in any given patient and, for certain products, that switching between products does not increase risk or diminish effectiveness.2
PK, PD and Immunogenicity Studies
Even when analytical similarity is well established, regulatory review often focuses closely on PK, PD and immunogenicity data.
Agencies expect bioanalytical methods that are:
- Scientifically justified and validated
- Sensitive and appropriately drug-tolerant
- Designed for direct biosimilar-to-reference comparison
- Fully documented and inspection-ready
PK similarity studies are highly sensitive to differences between products and are a central component of biosimilar review. Immunogenicity assessments—including antidrug antibody (ADA) and neutralizing antibody testing—are essential for evaluating safety and clinical comparability.
Design Considerations for Biosimilar Clinical Trials
Well-designed clinical studies are central to demonstrating similarity. Regulatory agencies expect sponsors to make scientifically justified design choices in several key areas.
- Study Population
Study populations should be selected to maximize sensitivity for detecting potential differences between the biosimilar and reference product. At the same time, the population must reflect relevant approved indications of the reference product. - Endpoints
Primary endpoints should be clinically meaningful and sensitive to differences between products. Sponsors must justify comparability margins statistically and demonstrate that these margins are appropriate for the therapeutic context. - Equivalence Versus Non-Inferiority
Most biosimilar trials are designed as equivalence studies rather than non-inferiority studies. Equivalence margins must be scientifically justified and supported by historical data for the reference product. - Extrapolation of Indications
If clinical similarity is demonstrated in one indication, regulators may allow extrapolation to other approved indications of the reference product without requiring additional clinical trials, provided there is adequate scientific justification.
Extrapolation decisions are based on the totality of evidence, including analytical similarity, mechanism of action, immunogenicity profile and clinical experience. - Post-Approval Requirements
Approval does not end regulatory oversight. Biosimilars may be subject to pharmacovigilance plans, risk management programs and additional studies for longterm safety monitoring.
The Modern Biosimilar Playbook: Reduce Uncertainty Early And Advance With Confidence
Leading biosimilar programs are structured to reduce regulatory uncertainty early through:
- Strategic reference product sourcing and comparability planning
- Head-to-head bioanalytical assay development
- High-throughput Phase I PK similarity execution
- Scientifically rigorous immunogenicity programs
- Submission-ready documentation aligned with FDA, EMA and ICH expectations
This approach supports efficient development timelines and reduces the risk of costly delays or additional studies.
Why Biosimilar Developers Choose Qps
Biosimilar success depends on analytical evidence. Regulators expect precise, defensible bioanalytical data that clearly demonstrate similarity to the reference product. QPS provides end-to-end biosimilar bioanalysis services across the full lifecycle aligned with regulatory guidance. These include:
Bioanalytical Assay Development and Validation
- PK assay development (Ligand Binding Assay [LBA] and LC-MS/MS)
- Head-to-head reference versus biosimilar assay design
- Good Laboratory Practice (GLP) validation aligned with regulatory guidance
- Cross-matrix and method robustness assessments
Phase I PK Similarity Bioanalysis
- High-throughput sample analysis
- Dense PK timepoint support
- Incurred sample reanalysis
- Similarity assessment-ready bioanalytical data packages
Immunogenicity Testing
- Tiered ADA assays
- Neutralizing antibody assays
- Cut point determination and drug tolerance optimization
- Longitudinal immunogenicity monitoring
Method Bridging and Analytical Comparability
- Platform and laboratory bridging studies
- Reference product lot to lot comparability
- Pre and post-manufacturing change bioanalysis
- Analytical justification for regulatory submissions
Late Stage and Confirmatory Biosimilar Bioanalysis
- PK/PD support for confirmatory studies
- Phase III bioanalysis (as required)
- Ongoing immunogenicity assessment
Regulatory Bioanalysis and Submission Support
- Bioanalytical sections for Investigational New Drug (IND), Biologics License Application (BLA) and Marketing Authorization Application (MAA) filings
- Responses to agency information requests
- Audit readiness and inspection support
- Scientific and statistical interpretation
Post Approval Biosimilar Bioanalysis and Lifecycle Management
- Comparability following scale-up or site changes
- Support for new presentations or devices
- Post-marketing and real-world evidence studies
- Ongoing immunogenicity surveillance
Your Partner for Global Biosimilar Programs
Biosimilar development is increasingly global. Sponsors often conduct multinational clinical programs, source reference products across regions and prepare submissions to multiple regulatory agencies simultaneously. Success depends on harmonized bioanalysis that meets the expectations of FDA, EMA and WHO without introducing unnecessary complexity or delay.
QPS bioanalysis teams are structured to support this reality. We bring experience across global regulatory frameworks and design bioanalytical strategies that remain consistent across regions, studies and lifecycle stages. Whether supporting multinational PK similarity trials or navigating cross-regional reference product sourcing, we help sponsors maintain alignment and momentum.
Key Takeaways
- Biosimilars are central to expanding patient access while reducing healthcare system costs.⁶
- Regulatory agencies rely on a totality-of-evidence framework that places significant weight on analytical and bioanalytical data.²
- The global biosimilars market is projected for strong growth through 2033.⁵
- Strong PD, PK and immunogenicity data are critical to regulatory confidence.
- QPS delivers regulator-ready bioanalysis to support global biosimilar approvals.
REFERENCES
- European Medicines Agency. Biosimilar medicines overview. European Medicines Agency. https://www.ema.europa.eu/en/human-regulatory-overview/
biosimilar-medicines-overview. Accessed 19 February 2026. - US Food and Drug Administration. Biosimilar review and approval. US Food and Drug Administration. https://www.fda.gov/drugs/biosimilars/reviewand-approval. Accessed 19 February 2026.
- US Food and Drug Administration. Biosimilar product information. US Food and Drug Administration. https://www.fda.gov/drugs/biosimilars/biosimilarproduct-information. Accessed 19 February 2026.
- Generics and Biosimilars Initiative (GaBI Online). Biosimilars approved in Europe. GaBI Online. https://www.gabionline.net/biosimilars/general/
biosimilars-approved-in-europe. Accessed 19 February 2026. - Grand View Research. Biosimilars market size, share & trends analysis report, 2025–2033. Grand View Research, 2025. https://www.grandviewresearch.
com/industry-analysis/biosimilars-market. Accessed 19 February 2026. - Association for Accessible Medicines. The U.S. Generic & Biosimilar Medicines Savings Report. Association for Accessible Medicines, 2025. https://
accessiblemeds.org/wp-content/uploads/2025/09/AAM-2025-Generic-Biosimilar-Medicines-Savings-Report-WEB.pdf. Accessed 19 February 2026.
